| USPEX 10.5 manual |
| USPEX 10.5 manual |
Suppose that the directory where the calculations are performed is  /StructurePrediction. This directory will contain:
/StructurePrediction. This directory will contain:
file INPUT.txt, thoroughly described in Section 4.
Subdirectory /StructurePrediction/Specific/ with VASP, SIESTA or GULP (etc.) executables, and enumerated input files for structure relaxation — INCAR_1, INCAR_2, …, and pseudopotentials.
Subdirectory /StructurePrediction/Seeds — contains files with seed structures and a list of compositions/anti-compositions. Seed structures should be in VASP5 POSCAR format and concatenated in a file called POSCARS or POSCARS_gen (gen is the generation number). The compositions and Anti-compositions files are used to control the compositions during variable-composition or single-block calculations.
Subdirectory /StructurePrediction/AntiSeeds — you may put here particular structures that you wish to penalize.
Executables and enumerated input files for structure relaxation (using external codes, like VASP, SIESTA, GULP, ...) should be put in subdirectory /StructurePrediction/Specific/
For VASP, put files INCAR_1, INCAR_2, …, etc., defining how relaxation and energy calculations will be performed at each stage of relaxation (we recommend at least 3 stages of relaxation), and the corresponding POTCAR_* files with pseudopotentials. E.g., INCAR_1 and INCAR_2 perform very crude structure relaxation of both atomic positions and cell parameters, keeping the volume fixed, INCAR_3 performs full structure relaxation under constant pressure with medium precision, INCAR_4 performs very accurate calculations. Each higher-level structure relaxation starts from the results of a lower-level optimization and improves them. POTCAR files of all relevant elements should also be in Specific/ folder, for instance POTCAR_C, POTCAR_O, etc.
For SIESTA, you need the pseudopotentials files and input files input_1.fdf,
input_2.fdf, …
For GULP, files goptions_1, goptions_2, …, and ginput_1, ginput_2, …must be present. The former specify what kind of optimization is performed, the latter specify the details (interatomic potentials, pressure, temperature, number of relaxation iterations, etc.).
For DMACRYS, fort.22 is the file for general control parameters. The classical force field is given by the file of fit.pots. File cutoff defines the maximum bond length of the intra-molecular bonds.
For CASTEP, structural files are given by cell_1, cell_2, …, while the computational parameters are given by param_1, param_2, …. The corresponding pseudopotential files must be present as well.
For CP2K, files cp2k_options_1, cp2k_options_2, …, must be present. All files should be normal CP2K input files with all parameters except atomic coordinates and cell parameters (these will be written by USPEX together with the finishing line “&END FORCE_EVAL”). The “name of the project” should always be USPEX, since the program reads the output from files USPEX-1.cell and USPEX-pos-1.xyz. We recommend performing relaxation at least in three steps (similarly to VASP) — first optimize only the atom positions with the lattice fixed, and then do a full relaxation.
For Quantum Espresso, files qEspresso_options_1, qEspresso_options_2, …, must be present. All files should be the normal QE input files with all parameters except atom coordinates, cell parameters and 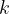 -points (these will be written by USPEX at the end of the file). We recommend performing a multi-step relaxation. For instance, qEspresso_options_1 does a crude structure relaxation of atomic positions with fixed cell parameters, qEspresso_options_2 does full structure relaxation under constant external pressure with medium precision; and qEspresso_options_3 does very accurate calculations.
-points (these will be written by USPEX at the end of the file). We recommend performing a multi-step relaxation. For instance, qEspresso_options_1 does a crude structure relaxation of atomic positions with fixed cell parameters, qEspresso_options_2 does full structure relaxation under constant external pressure with medium precision; and qEspresso_options_3 does very accurate calculations.
To use USPEX correctly, you should carefully edit the files in Specific/ folder to control the structure relaxation in USPEX. We take example of VASP as an external code:
Your final structures have to be well relaxed, and energies — precise. The point is that your energy ranking has to be correct (to check this, look at E_series.pdf file in the output).
Your POTCAR files: To yield correct results, the cores of your pseudopotentials (or PAW potentials) should not overlap by more than 10–15%.
To have accurate relaxation at low cost, use the multistage relaxation with at least three stages of relaxation for each structure, i.e. at least three INCAR files (INCAR_1, INCAR_2, INCAR_3, …). We usually set 4–5 stages of relaxation.
Your initial structures will be usually very far from local minima, in such cases it helps to relax atoms and cell shape at constant volume first (ISIF=4 in INCAR_1,2), then do full relaxation (ISIF=3 in INCAR_3,4), and finish with a very accurate single-point calculation (ISIF=2 and NSW=0 in INCAR_5).
Exceptions: when you do fixed-cell predictions, and also in evolutionary metadynamics (except full relaxation) you must have ISIF=2.
When your volume does not change, you can use default plane wave cutoff. When you optimize cell voluem (ISIF=3), you must increase it by 30–40%, otherwise you get a large Pulay stress. Also your convergence criteria can be loose in the beginning, but have to be tight in the end: e.g., EDIFF=1e-2 and EDIFFG=1e-1 in INCAR_1, gradually tightening to EDIFF=1e-4 and EDIFFG=1e-3 in INCAR_4. The maximum number of iterations (NSW) should be sufficiently large to enable good relaxation, but not too large to avoid wasting computer time on poor configurations. The larger your system, the larger NSW should be.
Choosing an efficient relaxation algorithm can save a lot of time. In VASP, we recommend to start relaxation with conjugate gradients (IBRION=2 and POTIM=0.02) and when the structure is closer to local minimum, switch to IBRION=1 and POTIM=0.3.
Even if you study an insulating system, many configurations that you will sample are going to be metallic, so to have well-converged results, you must use “metallic” treatment — which works both for metals and insulators. We recommend the Methfessel-Paxton smearing scheme (ISMEAR=1). For a clearly metallic system, use ISMEAR=1 and SIGMA=0.1–0.2. For a clearly insulating system, we recommend ISMEAR=1 and SIGMA starting at 0.1 (INCAR_1) and decreasing to 0.05.
Here we provide an example of INCAR files for carbon with 16 atoms in the unit cell, with default ENCUT=400 eV in POTCAR:
INCAR_1:
PREC=LOW
EDIFF=1e-2
EDIFFG=1e-1
NSW=65
ISIF=4
IBRION=2
POTIM=0.02
ISMEAR=1
SIGMA=0.10
INCAR_2:
PREC=NORMAL
EDIFF=1e-3
EDIFFG=1e-2
NSW=55
ISIF=4
IBRION=1
POTIM=0.30
ISMEAR=1
SIGMA=0.08
INCAR_3:
PREC=NORMAL
EDIFF=1e-3
EDIFFG=1e-2
ENCUT=520.0
NSW=65
ISIF=3
IBRION=2
POTIM=0.02
ISMEAR=1
SIGMA=0.07
INCAR_4:
PREC=NORMAL
EDIFF=1e-4
EDIFFG=1e-3
ENCUT=600.0
NSW=55
ISIF=3
IBRION=1
POTIM=0.30
ISMEAR=1
SIGMA=0.06
INCAR_5:
PREC=NORMAL
EDIFF=1e-4
EDIFFG=1e-3
ENCUT=600.0
NSW=0
ISIF=2
IBRION=2
POTIM=0.02
ISMEAR=1
SIGMA=0.05
The philosophy of input files for evolutionary metadynamics (calculationMethod=META) is very similar to USPEX, except that we DO NOT change the cell shape during the META evolution. Therefore, we need to put ISIF=2 for all META steps. If the full relaxation mode is on, we can put ISIF=3 for the steps of full relaxation. Therefore, if we have the following set up:
% abinitioCode 1 1 1 (1 1) % ENDabinit
the ISIF should be “2 2 2 3 3” for INCAR_1, …, INCAR_5 correspondingly.
Different from USPEX, VCNEB method doesn’t need structure relaxation from the external codes, and itself makes use of the forces completed by the external code. Take VASP INCAR files for example, we need to set NSW=0 to avoid the structure relaxation, but with ISIF=2 or 3 to extract the forces on the atoms, and the stress tensor in VASP. We also suggest to use PREC=Accurate to have a good estimation of the forces and stress for VCNEB. An example of INCAR file for VCNEB is presented below:
INCAR_1:
PREC=Accurate
EDIFF=1e-4
EDIFFG=1e-3
ENCUT=600.0
NSW=0
ISIF=2
IBRION=2
POTIM=0.02
ISMEAR=1
SIGMA=0.05
| USPEX 10.5 manual |