相同的结构可以再坐标系中以无限的方式展现出来(“模数不变性”)。 这些大量相同的选择将会导致非常扁平的晶胞单元,从而为结构弛豫和能量计算带来问题 (例如需要很大数量的 点)。约束条件在结晶学中已经熟知,那就是晶胞角度 在60
点)。约束条件在结晶学中已经熟知,那就是晶胞角度 在60 到120之间,不会移出任何一个多余的和有问题的晶胞 (例如:晶胞中允许的
到120之间,不会移出任何一个多余的和有问题的晶胞 (例如:晶胞中允许的 实际上是扁平的)。 因此我们发明了一种特殊的方法:用最短的晶格矢量来获得特殊的晶胞形状。如果至少有一个晶格矢量的投影在其他的晶格矢量 上或者在对立面上的斜的晶格矢量的长度大于(通过模比较)该晶格矢量长度的一半, 就会发生变形。也就是说,对于
实际上是扁平的)。 因此我们发明了一种特殊的方法:用最短的晶格矢量来获得特殊的晶胞形状。如果至少有一个晶格矢量的投影在其他的晶格矢量 上或者在对立面上的斜的晶格矢量的长度大于(通过模比较)该晶格矢量长度的一半, 就会发生变形。也就是说,对于 和
和 电子对, 或者是
电子对, 或者是 和
和 电子对, 有以下标准:
电子对, 有以下标准:
 |
 |
 |
(2) | ||
 |
|
 |
(3) | ||
 |
|
 |
(4) | ||
 |
|
 |
(5) |
例如,在标准2中,新的矢量 值等于:
值等于:
 |
(6) |
这种变形迭代进行,完全避免了劣质的晶胞形状,从而解决了问题。在该变形中, 原子坐标部分发生了改变,以保证初始结构和改变后的结构是完全相同的(在变形过程中, 原子的笛卡尔坐标保持不变)。
 variable minVectorLength
variable minVectorLength
Meaning: 设置最新产生的结构的晶胞参数的最小长度。
Default: 1.8  最大原子的共价直径。 对于分子晶体
最大原子的共价直径。 对于分子晶体
(calculationType = 310, 311) 默认最大值是 1.8 max(MolCenters).
Format:
2.0 : minVectorLength
当原子间距离足够小时,通常使用的计算方法(如赝势,PAW,LAPW和其他参数化的力场) 将不会起作用。这种情况需要避免,你可以用上三角形式的离子距离方阵 IonDistances 指定每对原子之间的最小距离:
variable IonDistances
Meaning: 在不同的原子类型之间设置最小原子距离矩阵。 小于离子距离的数值在物理意义上是没有意义的,需要严格避免。
Default: A和B原子之间的 离子距离被估计为 ,但是不会超过1.2
,但是不会超过1.2 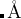 , 在分子类晶体计算中是
, 在分子类晶体计算中是 , 此处
, 此处 和
和 是USPEX中所估计得到的A原子和B原子的默认体积值。
是USPEX中所估计得到的A原子和B原子的默认体积值。
Format:
% IonDistances
1.0 1.0 0.8
0.0 1.0 0.8
0.0 0.0 1.0
% EndDistances
注意: 矩阵的大小必须等于原子种类数。如果上面例子中的化合物时 MgSiO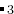 , 那么矩阵应该按如下解释:在一个新产生的结构中允许的Mg–Mg 距离为1.0 ,Mg–Si距离、Si–Si距离和O–O距离也是1.0 , 而最小的Mg–O距离和Si–O距离是0.8 。你可以把特定体系的更多 信息放进这个键长矩阵,举例来说,如果你知道Mg原子倾向离得很远,在你的体系中最近会 小于3 ,你可以列入该信息。然而需要注意的是最小距离越大,计算产生符合 这些约束条件的结构就越困难(特别是大体系)。所以尽量折中,并且谨记离子距离 必须远远小于实际的键长。
, 那么矩阵应该按如下解释:在一个新产生的结构中允许的Mg–Mg 距离为1.0 ,Mg–Si距离、Si–Si距离和O–O距离也是1.0 , 而最小的Mg–O距离和Si–O距离是0.8 。你可以把特定体系的更多 信息放进这个键长矩阵,举例来说,如果你知道Mg原子倾向离得很远,在你的体系中最近会 小于3 ,你可以列入该信息。然而需要注意的是最小距离越大,计算产生符合 这些约束条件的结构就越困难(特别是大体系)。所以尽量折中,并且谨记离子距离 必须远远小于实际的键长。
variable constraint_enhancement
Meaning: 可以用IonDistances矩阵(通过变为 constraint_enhancement的倍数)来严格约束计算产生的对称随机结构 (适用于所有变异操作,未增强的离子距离IonDistances矩阵依然可以应用)。 允许你应用离子距离矩阵中严格的约束条件(通过乘以增强约束条件)。 确保你在知道自己正在用的是什么意义的时候使用。
Default: 1
Format:
1 : constraint_enhancement
对分子晶体,下面的关键块体非常重要:
variable MolCenters
Meaning: 分子中心之间的最小距离矩阵。任何小于那些明确给出的分子 的大重叠的距离,在物理意义上都是不合理的,需要严格避免。
Default: 零矩阵适用于非分子计算, 分子晶体没有默认(必须明确给出)。
Format:
% MolCenters
5.5 7.7
0.0 9.7
% EndMol
注意: 在上面的例子中,有两种分子类型。在所有产生的结构中, 第一种类型的分子的几何中心之间的距离必须至少为5.5 (A–A距离), 第一种类型和第二种类型分子几何中心之间的距离— 7.7 (A-B距离), 第二种分子几何中心之间的距离— 9.7 (B–B距离)。