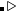 variable calculationMethod
variable calculationMethod
Meaning: 明确计算方法
可能值(特性):
USPEX— 晶体结构预测的发展算法
META—进化准动态算法
VCNEB — 用变晶胞微动弹性带方法探索过渡态路径
PSO — 修正的PSO算法
TPS — 过渡路径抽样方法(迄今未发布)
MINHOP — 最小值跳跃方法(迄今未发布)
COPEX — 另一种新技术,不久将发布
Default: USPEX
Format:
USPEX : calculationMethod
variable calculationType
Meaning:
也就是不论是体晶、纳米团簇还是表面结构,都可以被预测。 这种可变因素由三部分组成,包括 维度、 分子状态和 成分的可变性, 以及“s” or “S”表示的自旋选项:
维度:
“3” — 体材料
“2” — 表面, “–2” — 二维晶体
“1” — 聚合物
“0” — 纳米粒子
分子状态:
“0” — 无分子
“1” — 分子计算
?在计算中化学组成的变化:
“0” — 定成分
“1” — 变成分
magnetic calculation :
“s” or “S” – 设定磁性计算
Default: 300
Format:
301 : calculationType
注意: 如果calculationType=310,也就是,预测分子晶体,然后 USPEX希望你提供含有所有的分子类型的分子几何体的MOL_1, MOL_2, …这些分子将被安置在新的几何结构中。有效的选择:300(s300),301(s301), 310,000(s000),200(s200),201(s201),-200(-s200),(还有迄今未发布的:110,311)
variable optType
Meaning: 用户可以通过这个值来定义想要最大化或者最小化的性质。默认值是最小化焓和体积或者最大化optTypes剩下的性质设置-但是你可以清楚地定义你想要最大化或者最小化的性质。你也可以用某个目标值来优化性质(例如,对太阳能光伏来说带隙接近于1.34 eV的材料是很有吸引力的)。
可能的值 (特征):
Value |
Number |
Description |
enthalpy |
1 |
为了发现稳定相 |
volume |
2 |
体积最小化 |
(找到最密结构) |
||
hardness |
3 |
硬度最大化 |
(找到最硬相) |
||
struc_order |
4 |
有序度最大化 |
(发现最有序结构) |
||
aver_dist |
5 |
在一代内结构差别的平均值的最大化 |
diel_sus |
6 |
静态电介质极化率最大化 |
(仅适用于VASP和GULP) |
||
bandgap |
7 |
带隙最大化 |
(仅适用于VASP) |
||
diel_gap |
8 |
电能存储能量最大化 |
(仅适用于VASP) |
||
mag_moment |
9 |
磁化强度最大化 |
(仅适用于VASP) |
||
quasientropy |
10 |
结构准熵值最大化 |
与弹性力学相关的性质 (“11**”):
值 |
序号 |
说明 |
K, 体积弹性模量 |
1101 |
体弹性模量最大化 |
G, 剪切模量 |
1102 |
剪切模量最大化 |
E, 杨氏模量 |
1103 |
杨氏模量最大化 |
v, 泊松比 |
1104 |
泊松比最大化 |
G/K, Pugh模量比 |
1105 |
Pugh模量比最大化 |
Hv, 维氏硬度 |
1106 |
维氏硬度最大化 |
Kg, 断裂韧性 |
1107 |
断裂韧性最大化 |
D, 德拜温度 |
1108 |
德拜温度最大化 |
Vm, 声速 |
1109 |
声速最大化 |
S-波速 |
1110 |
S-波速最大化 |
P-波速 |
1111 |
P-波速最大化 |
注意: 与弹性力学相关的性质仅仅适用于VASP (从VASP5.1开始)和GULP。对于VASP用户, 需要在Specific/文件夹中增加至少一个INCAR_* 文件, INCAR_*文件应该包括的参数有IBRION=6, ISIF 3和 NFREE=4。 体积弹性模量剪切模量和杨氏模量的估计值是Voigh-Reuss-Hill(VRH) 的平均值。维氏硬度是用Chen-Niu模型 计算得到。 断裂韧性的优化用理论断裂韧性的最低值作为适用值。
3和 NFREE=4。 体积弹性模量剪切模量和杨氏模量的估计值是Voigh-Reuss-Hill(VRH) 的平均值。维氏硬度是用Chen-Niu模型 计算得到。 断裂韧性的优化用理论断裂韧性的最低值作为适用值。
Default: 焓值
Format:
enthalpy : optType(与Min_enthalpy这种方式设置的意义相同)
另外一个例子: Min_(bandgap-1.34)^2
需要注意的是在后一个例子里,我们优化的optType变量是一个数学表达式。数学表达式应该在圆括号内,同时应该是有效的MATLAB表达形式。完整的表达(包括 min_ and max_ 在内) 应该是一个整体,不要在整个表达式里填入空格。例如“( bandgap - 1.3 ) ^2"或者 min_ (bandgap-3)都是无效的形式。
注意: 如果optType=bandgap(带隙)或diel_gap(电介质带隙), 这里的带隙我们使用的是一个扩展函数,这个扩展函数对于金属表现出连续性。 —也就是说, 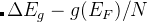 ,
,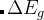 是带隙,
是带隙,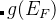 是在费米能级处 的态密度(对金属而言),而N是单胞内的原子数。由于这个扩展函数的连续性, 与带隙相关量的全局最大化甚至可以用于解决金属的问题。 对金属而言,这个值等于费米能级处的态密度。对于半导体和绝缘体而言这个值等于带隙。
是在费米能级处 的态密度(对金属而言),而N是单胞内的原子数。由于这个扩展函数的连续性, 与带隙相关量的全局最大化甚至可以用于解决金属的问题。 对金属而言,这个值等于费米能级处的态密度。对于半导体和绝缘体而言这个值等于带隙。
Fig. 5是以硬度为目标优化函数搜索TiO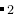 硬度最大的结构 (选择类型=硬度,显示了可能的最大硬度为14GPa, 这是对Dubrovinsky(2001)关于TiO的超硬度的反驳。 这是一个很好的例子:一个简单的USPEX就可以解决长期存在的争论。
硬度最大的结构 (选择类型=硬度,显示了可能的最大硬度为14GPa, 这是对Dubrovinsky(2001)关于TiO的超硬度的反驳。 这是一个很好的例子:一个简单的USPEX就可以解决长期存在的争论。
![\includegraphics[scale=0.18]{pic/hardness_example}](images/img-0040.png) .
. variable paretoRanking
Meaning: 打开/关闭帕累托排序方法,如果 paretoRanking 等于 1, USPEX 将用两种性质来对每一代的结构进行排序,Fitness 和能量在convex hull 之上(稳定性判断)。
可能的数值(整数)
0 — pareto ranking 方法没有被使用;
1 — pareto ranking 方法被使用。
Default: 0
Format:
1 : paretoRanking
现在,你需要明确所要计算体系的信息。
variable atomType
Meaning: 描述每种原子类型的特性。
Default: 无,必须明确给出
Format:
如果你更喜欢用门捷列夫元素周期表的原子序数,设置:
% atomTypeOr, if you prefer to use atomic names, specify:
% atomType
Mg Si O
% EndAtomType
或者,你更喜欢用元素名,设置:
% atomType
Magnesium Silicon Oxygen
% EndAtomType
variable numSpecies
Meaning: 描述每种类型原子的数目。
Default: 无,必须明确给出
Format:
% numSpecies
4 4 12
% EndNumSpecies
这表示第一种类型有4个原子,第二种类型有4个原子,第三种类型有12个原子。
Notes: 对于变成分的计算,你需要列出如下所示的组成构建块:
% numSpecies
2 0 3
0 1 1
% EndNumSpecies
这表示第一个构建块的分子式是AC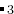 ,第二个构建块的分子式是BC, A、 B、 C在原子类型块中已经解释过。所有结构都满足公式
,第二个构建块的分子式是BC, A、 B、 C在原子类型块中已经解释过。所有结构都满足公式 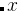 AC +
AC + 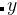 BC with , = (0,1,2,…) — 或者是A
BC with , = (0,1,2,…) — 或者是A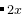 B
B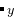 C
C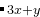 。如果你想做A-B-C体系中所有可能组成的预测,应该这样设置:
。如果你想做A-B-C体系中所有可能组成的预测,应该这样设置:
% numSpecies
1 0 0
0 1 0
0 0 1
% EndNumSpecies
你也可以用一个化学式的计量比数来做定成分计算,这种情况下, 只需要设置一行(calculationType=301)如下为ABC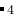 化合物的例子:
化合物的例子:
% numSpecies
2 1 4
% EndNumSpecies
variable magRatio
Meaning: 对非自旋极化(NM),FM-LS、FM-HS、AFM-L、AFM-H、FM-LH和 AF-LH所对应的不同类型磁性质的结构的初始比例。仅VASP支持此功能
Default: 0.1, 0.9/4, 0.9/4, 0.9/4, 0.9/4, 0, 0
Format:
% magRatio
1/8 1/8 1/8 1/8 1/8 0 0
% EndMagRatio
这意味着产生NM、FM-LS、FM-HS、AFM-L和 AFM-H结构的比率均为20%(这里不是1/8,已经重新调整为1)。 没有FM-LH和AF-LH磁化态的结构产生。
Notes:
(1)总的来说,磁化率magRatio可以大于1,其比率将会自动调整为1。 (2)在USPEX中的含义和初始磁化率的值:
NM — 非自旋极化;
FM-LS — 低自旋铁磁;
FM-HS — 高自旋铁磁;
AFM-L — 低自旋反铁磁;
AFM-H — 高自旋反铁磁;
FM-LH — 低/高自旋混合铁磁;
AF-LH — ?低/高自旋混合反铁磁。
(3) 对HM(非自旋极化),所有原子的初始磁化值均为0。对自旋状态和高自旋状 态原子的初始磁化值分别可以设置为MAGMOM = 1和4。对低/高自旋混合状态,每个 原子的磁化值可以被随意地设置为MAGMOM = 1或4。
(4) 当单胞中有奇数个原子时,将不会产生AFM类型结构。
(5) 磁化率magRatio亦适用于在自旋突变操作中的突变值。
variable ldaU
Meaning: 使用LDA+U方法,指定每种原子的Hubbard U值。仅VASP支持此功能
Default: 0 对应每种类型的原子
Format:
% ldaU
4 0
% EndLdaU
variable ExternalPressure
Meaning: 指定你所要计算体系的外部压力,单位是GPa。
Default: 0
Format:
100 : ExternalPressure
Note: 从 USPEX 9.4.1 起,压力的值(单位 GPa)可以直接在INPUT.txt文件中设置。不需要再 弛豫优化文件Specific/再次指定。
variable valences
Meaning: 描述每类原子的化合价。 这仅用于评估结合的硬度,而结合硬度又用来计算近似的动力学矩阵(对软模变异) 和晶体的硬度。
Default: USPEX有一系列的系统默认化合价 (见附录 9.8). 然而,需要注意的是,对一些元素 (例如N、S、W、Fe和 Cr等),有许多可能的化合价。除非你要计算硬度, 否则这没有问题,你可以用系统默认化合价。如果你要做硬度计算,你必须明确给出化合价。
Format:
% valences
2 4 2
% EndValences
variable goodBonds
Meaning: 在矩阵中指定两原子间最低的键价被认为是最重要的键价。 就像离子距离矩阵IonDistances (如下),这是一个上三角形式的方阵。这仅用来计算硬度和软模变异。 我们可以用公式 goodBonds=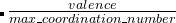 。
。
Default: USPEX可以给出goodBonds合理的默认估值,你可以在输出文件OUTPUT.txt 中得到这些数值。对大多数情况,用默认值已经足够,但是对于硬度的计算,你必须要仔细地审查这些数值, 或者手动设置。更多细节,请参考附录 9.9
Format:
% goodBonds
10.0 10.0 0.2
0.0 10.0 0.5
0.0 0.0 10.0
% EndGoodBonds
注意: 矩阵的维度必须与原子种类数或原子名称数相等。如果只有一种原子,那么矩阵就填充该数字。 上面的矩阵解读如下:可被认为是一个键,Mg–Mg间的距离要足够短使其键价为10或者更大, Mg-Mg键的距离必须足够短。对Mg–Si键,Si–Si键和O–O键也有同样要求(通过使用这种 专一的标准,我们有效地排除了来自软模变异和硬度计算的交互作用), 然而在用于硬度和软模变异计算的Mg-O键的键价取为0.2或者更大,Si–O键的键价取为 0.5或者更大。
variable checkMolecules
Meaning: 打开或关闭原始分子的再弛豫 (MOL_1, MOL_2, …)是否完整。可用于分子晶体 (calculationType=310, 311)。
可能的数值(整数):
0 — 未经核实,被认为是损坏了的结构或是合并的分子(我们强烈建议不使用这种类型)。
1 — 执行检查命令,所有损坏或是合并的分子均被遗弃。
Default: 1
Format:
1 : 核实分子
variable fitLimit
Meaning: 对于最大值: 如果所获得fitness的最小值比fitLimit的值小, 那么USPEX的计算将会在第一代之后结束。 对于最小值: 如果所获得fitness的最小值比 fitLimit的值大,那么USPEX的计算将会在第一代之后结束。
Default: 无默认值,用户需要自己设定。
Format:
10 : fitLimit