| USPEX 10.2 manual |
| USPEX 10.2 manual |
EX01-3D_Si_vasp: Silicon (8 atoms/cell) at zero pressure. Variable-cell DFT calculation using VASP, PBE96 functional. Many thanks to G. Kresse for permission to include his PAW files (POTCAR) in our distribution.
EX02-3D_MgAl2O4_gulp: MgAl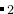 O
O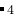 (28 atoms/cell) at 100 GPa pressure. Variable-cell calculation using Buckingham potentials, GULP code. Beware that for reliable results, you should better do ab initio calculations.
(28 atoms/cell) at 100 GPa pressure. Variable-cell calculation using Buckingham potentials, GULP code. Beware that for reliable results, you should better do ab initio calculations.
EX03-3D-const_cell_MgSiO3_gulp: this example shows how to do structure prediction when you know cell parameters. MgSiO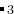 (20 atoms/cell) with Buckingham potentials, GULP code. Cell parameters correspond to post-perovskite. The discovery of post-perovskite (Oganov & Ono, Nature 2004; Murakami et al., Science 2004) was a major breakthrough in Earth sciences.
(20 atoms/cell) with Buckingham potentials, GULP code. Cell parameters correspond to post-perovskite. The discovery of post-perovskite (Oganov & Ono, Nature 2004; Murakami et al., Science 2004) was a major breakthrough in Earth sciences.
EX04-3D_C_lammps: this example shows how to do crystal structure prediction using USPEX together with the LAMMPS code. In this a simple example: 8 carbon atoms, and Tersoff potential.
EX05-3D_Si_atk: Example of crystal structure prediction of Si with 8 atoms/cell using the density-functional tight binding approximation and ATK code.
EX06-3D_C_castep: DFT-based prediction of the crystal structure of carbon with 8 atoms/cell at 10 GPa, using the CASTEP code.
EX07-2D_Si_vasp: prediction of the 2D-crystal of silicon using DFT and VASP. Simple and powerful.
EX08-0D_LJ_gulp: Nanoparticle structure prediction. Lennard-Jones nanoparticle with 30 atoms, using the GULP code.
EX09-3D-molecules_CH4_vasp: methane with 4 molecules/cell, at the pressure of 20 GPa. DFT, VASP. Molecule is described in the file MOL_1.
EX10-3D-molecules_CH4_dmacrys: methane with 8 molecules/cell, with forcefield and DMACRYS code, at normal pressure. Molecule is described in the file MOL_1, but note its slightly unusual format for DMACRYS calculations. Please put executables dmacrys, neighcrys-pp, neighcrys-vv in the Specific/ folder.
EX11-3D-molecules_urea_tinker: urea with 2 molecules/cell, with forcefield and TINKER code, at normal pressure. Molecule is described in the file MOL_1.
EX12-3D_varcomp_LJ_gulp: Lennard-Jones binary system with fake “Mo” and “B” atoms, GULP, and variable-composition USPEX (Lyakhov and Oganov, 2010).
EX13-3D_special_quasirandom_structure_TiCoO: USPEX can easily find the most disordered (or the most ordered) alloy structure. Here, this is shown for Ti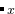 Co
Co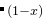 O. You need to specify the initial structure in Seeds/POSCARS and use only the permutation operator. In this case, you don’t need to use any external codes. In this example, we optimize (minimize) the structural order (Oganov and Valle (2009); Lyakhov, Oganov, Valle (2010)) without relaxation (abinitioCode = 0). Seed structure (supercell of Ti-Co-O-structure) is permutated to find the structure the minimum/maximum order. Minimizing order in this situation, one gets a generalized version of the “special quasirandom structure”.
O. You need to specify the initial structure in Seeds/POSCARS and use only the permutation operator. In this case, you don’t need to use any external codes. In this example, we optimize (minimize) the structural order (Oganov and Valle (2009); Lyakhov, Oganov, Valle (2010)) without relaxation (abinitioCode = 0). Seed structure (supercell of Ti-Co-O-structure) is permutated to find the structure the minimum/maximum order. Minimizing order in this situation, one gets a generalized version of the “special quasirandom structure”.
EX14-GeneralizedMetadynamics_Si_vasp: simple example of a powerful capability to find complex low-energy structures starting with a simple seed structure (Zhu et al, 2013). Silicon, up to 16 atoms/cell, DFT, VASP. Pay special attention to INCAR files. Best of all, just keep the files that you see here, changing only ENCUT, perhaps SIGMA. Evolutionary metadynamics not only predicts low-energy structures, but also gives an idea of transition mechanisms between crystal structures.
EX15-VCNEB_Ar_gulp: example of a variable-cell nudged elastic band (VCNEB: Qian et al., 2013) calculation of the fcc-hcp transition in a model system, argon, at 0 GPa pressure. Lennard-Jones potential, GULP code.
EX16-USPEX-performance_SrTiO3_gulp: SrTiO (50 atoms/cell) at zero pressure. Variable-cell calculation using Buckingham potentials, GULP code. Running this example you can see that even for such a relatively large system USPEX code scores a  90% success rate and remarkable efficiency. This contrasts with a 7-12% success rate reported for the same system and using the same potential by Zurek & Lonie. Clearly, USPEX outperforms the poor reimplementation of our method by Zurek and Lonie. We have witnessed excellent performance of our code also for much larger systems.
90% success rate and remarkable efficiency. This contrasts with a 7-12% success rate reported for the same system and using the same potential by Zurek & Lonie. Clearly, USPEX outperforms the poor reimplementation of our method by Zurek and Lonie. We have witnessed excellent performance of our code also for much larger systems.
EX17-3D_DebyeTemp_C_vasp: example of optimization of the elasticity-related properties (bulk or shear moduli, Poisson ratio, Chen-Niu hardness, or Debye temperature). In this example, we maximize the Debye temperature of carbon using the VASP code.
EX18-3D_varcomp_ZnOH_gulp: as you know, USPEX has unique capabilities for variable-composition searches. This example shows a pretty challenging case — variable-composition calculation for the ternary system Zn-O-H. This calculation uses a ReaxFF forcefield in GULP code. USPEX can do calculations for any number of components — e.g. quaternary, quinternary, etc. systems are within its reach. Of course, the more components you have, the more expensive (and the more risky) your calculation is. No reference results at the moment.
EX19-Surface-boron111: Prediction of (111) surface reconstruction of alpha-boron, with variable number of atoms (Zhou et al., Phys. Rev. Lett. 113, 176101 (2014)).
EX20-0D_Cluster_C60_MOPAC: Cluster structure prediction (000) for C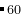 using MOPAC.
using MOPAC.
EX21-META_MgO_gulp: Evolutionary metadynamics, with GULP code and Buckingham potentials, MgO with 8 atoms/cell. Starting structure is of rocksalt type, and evolutionary metadynamics finds a number of low-energy structures and structural relations.
EX22-GEM_MgO_gulp: Generalized evolutionary metadynamics, with GULP code and Buckingham potentials. Starting structure is of rocksalt type, with 8 atoms/cell, the calculation is allowed to increase system size up to 16 atoms/cell, and generalized evolutionary metadynamics (GEM) finds a number of low-energy structures and structural relations.
EX23-MgO_surface: Prediction of surface reconstruction of magnesium-oxygen, with fixed number of atoms.
EX24-SingleBlock_Magnetic_Fe3C_VASP: Magnetic structure prediction (300) for Fe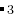 C in single block using VASP.
C in single block using VASP.
EX25-3D-C8-DFTB: Structure prediction (300) for C (8 atoms/cell) using DFTB+ with 3ob-3-1 set. The energy differnet between graphite and diamond seems to be overestimated by the current DFTB parameter set.
EX26-Ar_TPS: TPS calculation for the phase tranformation in Ar hcp and fcc solid of 8000 atoms at 40K under 1 atmosphere.
EX27-3D-P2_FHIaims: Structure prediction (300) for P (2 atoms/cell) using FHI-aims.
EX28-0D-Cluster_Cu9_FHIaims: Cluster structure prediction (000) for Cu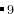 using FHI-aims.
using FHI-aims.
EX29-Si_gap-maximize_singleblock: Structure prediction (300) of silicon with optimization of the band gap using a meta-GGA functional.
EX30_Pd_111-oxidation: Reconstructions involving foreign species on elemental surfaces (such as PdO@Pd(111) surface).
EX31_2D_varcomp_SnS_VASP: Variable composition search for stable 2D-crystal (layer) Sn-S phases. Example is provided by Z.A.
EX32_pmpaths: Generating by the pmpaths 5 different pathways of diamond-graphite transition. Example is provided by V. Stevanovic.
EX33-carbon_QE: Fixed-composition search for the lowest-energy crystal structure with 8 atoms/cell (300), using Quantum Espresso.
| USPEX 10.2 manual |