| USPEX 10.5 manual |
| USPEX 10.5 manual |
For a molecular crystal, the MOL_1 file describes the internal geometry of the molecule from which the structure is built. The Z_Matrix file is created using the information given in the MOL_1 file, i.e., bond lengths and all necessary angles are calculated from the Cartesian coordinates. The lengths and angles that are important should be used for the creation of Z_Matrix — this is exactly what columns 5–7 specify. Let’s look at the MOL_1 file for benzene C H:
H:
![\includegraphics[scale=0.6]{pic/benzene.png}](images/img-0105.png)
The 1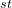 atom is H, its coordinates are defined without reference to other atoms (“0 0 0”).
atom is H, its coordinates are defined without reference to other atoms (“0 0 0”).
The 2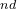 atom is C, its coordinates (in molecular coordinate frame) in Z_matrix will be set only by its distance from the 1 atom (i.e. H described above), but no angles — (“1 0 0”).
atom is C, its coordinates (in molecular coordinate frame) in Z_matrix will be set only by its distance from the 1 atom (i.e. H described above), but no angles — (“1 0 0”).
The 3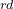 atom is C, its coordinates will be set by its distance from the 2 atom, and the bond angle 3-2-1, but not by torsion angle — hence we use “2 1 0”.
atom is C, its coordinates will be set by its distance from the 2 atom, and the bond angle 3-2-1, but not by torsion angle — hence we use “2 1 0”.
The 4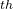 atom is C, its coordinates will be set by its distance from the 3 atom, bond angle 4-3-2, and torsion angle 4-3-2-1 — hence, we use “3 2 1” and so forth…until we reach the final, 12 atom, which is H, defined by its distance from the 7 atom (C), bond angle 12-7-6 and torsion angle 12-7-6-11 — hence “7-6-11”.
atom is C, its coordinates will be set by its distance from the 3 atom, bond angle 4-3-2, and torsion angle 4-3-2-1 — hence, we use “3 2 1” and so forth…until we reach the final, 12 atom, which is H, defined by its distance from the 7 atom (C), bond angle 12-7-6 and torsion angle 12-7-6-11 — hence “7-6-11”.
The final column is the flexibility flag for the torsion angle. For example, in C4, the tosion angle is defined by 4-3-2-1. This flag should be 1 for the first three atoms, and 0 — for the others, if the molecule is rigid. If any other flexible torsion angle exists, specify 1 for this column.
For polymers, the MOL_1 file is used to represent the geometry of a monomeric unit, in the same style as for molecular crystals, except that we use the last column to specify the reactive atoms as shown in the MOL_1 file for PVDF:
![\includegraphics[scale=0.72]{pic/PVDF_MOL.png}](images/img-0109.png)
Above we described a generic MOL_1 file format. However, some classical forcefield based codes need additional information. For instance, GULP needs to specify the chemical labels and charge. The MOL_1 file for aspirin can be written in the following way:
Aspirin_charge Number of atoms: 21 H_1 0.2310 3.5173 4.8778 0 0 0 1 0.412884 O_R 0.7821 4.3219 4.9649 1 0 0 1 -0.676228 C_R 0.4427 5.0883 6.0081 2 1 0 1 0.558537 O_2 -0.5272 4.5691 6.6020 3 2 1 0 -0.658770 C_R 1.0228 6.3146 6.3896 3 2 4 0 0.116677 C_R 2.1330 6.8588 5.6931 5 3 2 0 0.311483 C_R 0.4810 7.0546 7.4740 5 3 6 0 -0.119320 O_R 2.8023 6.2292 4.6938 6 5 3 0 -0.574557 C_R 2.6211 8.1356 6.0277 6 5 8 0 -0.083091 C_R 0.9966 8.3146 7.8237 7 5 3 0 -0.103442 H_2 -0.3083 6.6848 8.0128 7 5 10 0 0.198534 C_R 3.6352 5.1872 4.9079 8 6 5 0 0.609295 C_R 2.0623 8.8613 7.0940 9 6 5 0 -0.119297 H_2 3.3963 8.5283 5.4906 9 6 13 0 0.174332 H_2 0.5866 8.8412 8.6013 10 7 13 0 0.205960 O_2 3.9094 4.7941 6.0632 12 8 6 0 -0.588433 C_3 4.2281 4.5327 3.7638 12 8 16 0 -0.271542 H_2 2.4227 9.7890 7.3367 13 9 10 0 0.196738 H_2 3.4269 4.1906 3.1183 17 12 8 0 0.151315 H_2 4.8283 3.6848 4.0792 17 12 19 0 0.131198 H_2 4.8498 5.2464 3.2337 17 12 19 0 0.127726
Here, the keyword charge in the title tells the program to read the charge in the additional (last) column.
To work with Tinker, the additional column must specify the atomic type label as follows:
Urea Number of atoms: 8 C 0.000000 0.000000 0.000000 0 0 0 1 189 O 0.000000 0.000000 1.214915 1 0 0 1 190 N 1.137403 0.000000 -0.685090 1 2 0 1 191 N -1.137403 0.000000 -0.685090 1 2 3 0 191 H 1.194247 0.000000 -1.683663 4 1 3 0 192 H -1.194247 0.000000 -1.683663 4 1 3 0 192 H 1.998063 0.000000 -0.138116 2 1 3 0 192 H -1.998063 0.000000 -0.138116 2 1 3 0 192
There are plenty of programs which can generate Zmatrix style files, such as Molden, Avogadro, and so on. Experienced users might have their own way to prepare these files. For the users’ convenience, we have created an online utility to allow one to generate the USPEX-style MOL file just from a file in XYZ format. Please try this utility at bluehttps://uspex-team.org/online_utilities/zmatrix/.
| USPEX 10.5 manual |