EX01-3D_Si_vasp: 零压力下的硅(每个单胞中含8个原子)。使用VASP, PBE96 泛函进行变胞 DFT计算。感谢G. Kresse和他的PAW文件(POTCAR)对我们的贡献。
EX02-3D_MgAl2O4_gulp: 在100 GPa 下的MgAl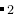 O
O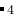 (每一个单胞 中有28个原子)。使用Buckingham势,GULP代码进行变胞计算。如果需要很可靠的结果,最好用ab initio算法。
(每一个单胞 中有28个原子)。使用Buckingham势,GULP代码进行变胞计算。如果需要很可靠的结果,最好用ab initio算法。
EX03-3D-const_cell_MgSiO3_gulp:这个例子向我们展示在知道单胞 参数时如何进行结构预测。含有Buckingham势,GULP 代码的MgSiO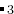 (每个单胞有20个原子)。 晶胞参数与后钙钛矿一致。后钙钛矿的发现(Oganov & Ono, Nature 2004; Murakami et al., Science 2004)在地球科学领域是一个重大突破。
(每个单胞有20个原子)。 晶胞参数与后钙钛矿一致。后钙钛矿的发现(Oganov & Ono, Nature 2004; Murakami et al., Science 2004)在地球科学领域是一个重大突破。
EX04-3D_C_lammps:这个例子展示了如何使用结合了LAMMPS码的USPEX进行 晶体结构预测。在这个简单的例子中:有8个碳原子,采用Tersoff势。
EX05-3D_Si_atk:这是一个使用密度泛函紧束缚近似和ATK 代码对每一单 胞含8个原子的硅进行晶体结构预测的例子。
EX06-3D_C_castep:在0 GPa下,使用CASTEP代码对每个单胞含8个原子的 碳的晶体结构进行基于DFT的预测。
EX07-2D_Si_vasp: 使用DFT和VASP预测硅的二维晶体。简单而有效。
EX08-0D_LJ_gulp:纳米结构预测。含有30个原子的Lenard-Jones纳米颗粒, GULP代码。
EX09-3D-molecules_CH4_vasp:甲烷分子晶体预测,压力20 GPa,DFT, VASP。分子在文件MOL_1中描述。
EX10-3D-molecules_CH4_dmacrys:常压,带有8个分子的单胞,采用力场 和DMACRYS代码。在文件MOL_1中描述分子,但要注意其DMACRYS计算略有不同的格式。 请把可执行文件dmacrys、neighcrys-pp和neighcrys-vv放在 文件夹Specific/。
EX11-3D-molecules_urea_tinker:常压,2个分子的单胞,采用力场和 TINKER代码。在文件MOL_1中描述分子。
EX13-3D_special_quasirandom_structure_TiCoO: 含有伪“Mo”和“B”原子,GULP和变组成 USPEX(Lyakhov and Oganov, 2010)的Lenard-Jones二元体系。
EX13-3D_special_quasirandom_structure_TiCoO:USPEX可以很容易 地找到最无序(或最有序)合金结构。这里表示为Ti Co
Co O。你需要在Seeds/POSCARS 指定初始结构和仅使用置换操作。在这种情况下,不需要使用任何外部代码。在这个例子中, 我们在无弛豫的情况下(abinitioCode = 0)优化(最小化)结构有序度(Oganov and Valle (2009); Lyakhov, Oganov, Valle (2010))。 种子结构(Ti-Co-O结构的超晶胞)被置换找到结构的最小/最大化有序度。在这种情况下最小化有序度, 可以得到“特殊准随机结构”的通用版本。
O。你需要在Seeds/POSCARS 指定初始结构和仅使用置换操作。在这种情况下,不需要使用任何外部代码。在这个例子中, 我们在无弛豫的情况下(abinitioCode = 0)优化(最小化)结构有序度(Oganov and Valle (2009); Lyakhov, Oganov, Valle (2010))。 种子结构(Ti-Co-O结构的超晶胞)被置换找到结构的最小/最大化有序度。在这种情况下最小化有序度, 可以得到“特殊准随机结构”的通用版本。
EX14-GeneralizedMetadynamics_Si_vasp:关于开始用一个简单的种子结构搜索复杂 低能量结构的简单例子(Zhu et al, 2013)。硅,最多为16个原子的单胞,DFT,VASP。 要特别注意到INCAR文件。最重要的是,只要保留你在这里看到的文件,只 改变ENCUT,SIGMA,进化准动力学不仅预测低能量的结构,同时也给出 了晶体结构之间的转换机制。
EX15-VCNEB_Ar_gulp:氩气,在0 GPa压力模型系统中采用变胞微动弹性 带(VCNEB: Qian et al., 2013)计算fcc-hcp转变。Lenard-Jones势,GULP代码。
EX16-USPEX-performance_SrTiO3_gulp: SrTiO(50原子/单胞)在零压力下。 使用Buckingham势,GULP代码进行可变单胞计算。从这个例子中可以看到,即使是这样一个比较大 的系统,USPEX代码仍有 90%的成功率和显著的效率。与此相反,通过Zurek和Lonie使用相同的体系, 相同的势能报道的成功率只有7-12%。显然,USPEX优于我们通过Zurek和Lonie的实现方法。 我们已经见证了我们的代码的性能在更大的系统也很优异。
90%的成功率和显著的效率。与此相反,通过Zurek和Lonie使用相同的体系, 相同的势能报道的成功率只有7-12%。显然,USPEX优于我们通过Zurek和Lonie的实现方法。 我们已经见证了我们的代码的性能在更大的系统也很优异。
EX17-3D_DebyeTemp_C_vasp: 对弹性相关性能优化的例子(体积和剪切模量, 泊松比,Chen-Niu硬度,德拜温度)在这个例子中,我们使用VASP代码最大限度地提高碳的德拜温度。
EX18-3D_varcomp_ZnOH_gulp:正如你所知道的,USPEX对于变成分的搜索 具有独特的能力。这个例子为我们展现了一个非常具有挑战性的情况—对于三元体系Zn-O-H的变成分 计算。这个计算在GULP代码中使用ReaxFF力场。USPEX可以为任何成分组成进行计算–例如,在其它 范围内可实现的四元和五元体系。当然,成分越多,计算越昂贵(和越有风险)。目前没有任何参考结果。
EX19-Surface-boron111: 预测alpha-B的(111)表面重构,采用变原子数(Zhou et al., Phys. Rev. Lett. 113, 176101 (2014)).
EX20-MinHop_SiO2_gulp: SiO (300)的最小跳跃法计算。
(300)的最小跳跃法计算。
EX21-SingleBlock_Magnetic_Fe3C_VASP: 在单块模式(300)中使用VASP预测 Fe C磁性结构的预测。
C磁性结构的预测。
EX22-3D_Cluster_C60_MOPAC: 使用MOPAC进行C 团簇结构的预测(000)。
团簇结构的预测(000)。
EX23-META_MgO_gulp: 进化准动力学方法,采用GULP代码和Buckingham势, 用含有8个原子的MgO初始结构是盐岩型结构,进化准动力学方法找到许多能量低的结构和结构之 间的关系。
EX24-GEM_MgO_gulp: 通用进化准动力学方法,采用GULP代码和Buckingham势, 用含有8个原子的单胞,这个计算是允许增加到16个原子,通用进化准动力学方法找到许多能量低 的结构和结构之间的关系。