5. Input options. The input.uspex file¶
The input.uspex file has json-like syntax and hierarchical structure representing
modular nature of USPEX program. Below we
describe the most important parameters of the input. Most of the
parameters have reliable default values (this allows you to have
extremely short input files!). Those options that have no default should
always be specified. Please consult online utilities at
https://uspex-team.org/online_utilities/ — these help to prepare input
and analyze some of results. Section 6 of
this Manual briefly discusses these utilities.
In order to make structure of parameters and blocks of the input.uspex file
more visual,
we have prepared an example code template, which include every block and parameter.
You can see it in Section 7.1 .
5.1. The input.uspex file syntax¶
The input.uspex consists of main section and a number of definition sections.
Main section is mandatory and precedes definition sections. Definition
sections are optional. There could be any number of definition sections.
Each definition section starts with define name line.
{...}
#define name1
{...}
#define name2
{...}
#define name3
{...}
...
Main input section is described in Section 5.2, Section 5.3, Section 5.4, Section 5.5 and Section 5.6.
Definition sections correspond to parameters of ab initio calculations (Section 5.7), submission parameters (Section 5.8), molecule definitions (Section 5.9) and environment definitions (Section 5.10).
Each section is a dictionary of key, value pairs.
{
key1 : value1
key2 : value2
key3 : value3
...
}
Such pairs correspond to different parameters or blocks of parameters. Input parameters are grouped into blocks recursively. Each block reflects some aspect of calculation. So it should be quite intuitive to prepare such input file.
Parameters can have numeric, string, sequence or map values.
numGenerations : 50,
stages: [vasp1 vasp2 vasp3]
ionDistances: {'C C': 2.0 'C H': 1.2 'H H': 0.7}
Blocks of parameters look as follows:
compositionSpace: {
symbols: ...
blocks: ...
}
There are two types of sequences: lists [] and tuples ().
Tuples are expected to have fixed length when lists can have arbitrary one.
5.2. General calculation parameters¶
The most general algorithm used in all USPEX modes is iteration. At each
iteration step program deals with a number of systems also called
individuals. And the set of these systems is called population or
generation. Such iteration is controlled by two parameters: numGenerations and
stopCrit.
numGenerations:Maximum number of generations allowed for the simulation. The simulation can terminate earlier, if the same best structure remains unchanged for
stopCritgenerations.Default:
Format:
numGenerations : 50
stopCrit:The simulation is stopped if the best structure did not change for
stopCritgenerations, or when numGenerations have expired – whichever happens first.Default:
Format:
stopCrit : 20
USPEX job with population consists of two parts: creation and relaxation.
Creation is controlled by block optimizer. This block depends on the type
of optimization. For global optimum search it is
GlobalOptimizer. In later releases VCNEB optimizer type
will be available for optimization of a single phase transition pathway.
optimizer:Specifies the type of calculation and its parameters
- Available types:
GlobalOptimizer| global search algorithm see Section 5.3.Default:
Format:
optimizer : {type: GlobalOptimizer ...}
For population relaxation USPEX employs a powerful two-level parallelization scheme, making its parallel scalability exemplary. The first level of parallelization is performed within structure relaxation codes, the second level of parallelization distributes the calculation over the individuals in the same population (since structures within the same generation are independent of each other).
stages:List of relaxation stages to be applied to each individual in population. The names of definition sections should be used in this sequence. See Section 5.7 for information on writing this section.
Default:
Format:
stages : [gulp gulp gulp]
numParallelCalcs:Specifies how many structure relaxations you want to run in parallel.
Default:
Format:
numParallelCalcs : 10
One may specify output file according to their convenience.
output:Specifies the content and title of the columns in the output. These columns will be used whenever details of an individual will be printed (
Individuals,goodStructures, etc.).Default: depends on calculation type
Format:
output: { columns: [ (simpleMoleculeUtility.composition 'Composition') (radialDistributionUtility.structureOrder 'Structure order') (radialDistributionUtility.averageOrder 'Average order') (radialDistributionUtility.quasientropy 'Quasientropy')] }
outputRefreshDelay:Time (in seconds) between file updates in the results folder
Default: 120
Format:
outputRefreshDelay : 30
5.3. Global optimization search¶
Global optimization is one of possible types of calculation in USPEX (see
optimizer in Section 5.2). As the title suggests it performs
search for global optimum of some property over certain configuration
space. Such search is done by sampling this space iteration by
iteration. Sample of this space is called population. Target
configuration space is described with parameters block target,
property to be optimized with optType parameter and sampling
method with selection block.
{
type: GlobalOptimizer
target: {...}
selection: {...}
optType: ...
}
target:Specifies the target space of search
Default:
- Available types:
Atomistic– structure prediction (see Section 5.5). This target covers all regimes which deals with structures consisting of atoms including molecular crystals, various dimensions and environments (like substrate). Use case examples: crystals (atomic and molecular), thin films, surfaces, nanoparticles etc.Format:
target : {type: Atomistic ...}
selection:Specifies the evolutionary algorithm to be used
Default:
- Available algorithms:
USPEXClassic– full evolutionary algorithm (see Section 5.6).Format:
selection : {type: USPEXClassic ...}
optType:This parameter specifies the property (or properties) that you want to optimize (see Section 5.4)
Default:
Format:
optType : enthalpy
5.4. Optimization type¶
Optimization type specification is designed as powerful method of writing functions inside input file. Such functions looks like
(func arg1 arg2 ...)
Here arg1, arg2 could be functions themselves. So this
procedure could be recursive. For example:
(pareto (aging enthalpy) (negate radialDistributionUtility.structureOrder))
This means that parameter to be optimized will be calculated for each
structure by following procedure. Enthalpy will be taken from structutre
directly, parameter structureOrder will be calculated using
radialDistributionUtility module. Then aging function
will be applied to enthalpy and negate function to
structureOrder. Then finally optimization parameter will be
determined via pareto function with two arguments: aged enthalpy
and negated structure order.
For now there are following functions available:
Function |
Description |
|---|---|
|
takes opposite value |
|
apply penalties to old structures (not tunable at current release) |
|
calculate Pareto front in space of given arguments. |
Available base parameters:
Name |
Description |
|---|---|
|
enthalpy of system |
|
enthalpy per block above convex hull (for variable composition) |
|
volume of system unit cell |
|
degree of order |
|
structural quasientropy |
Providing only base parameter as optType is also a valid option.
5.5. Crystal structure prediction¶
When target type in Section 5.3 is set to
Atomistic USPEX performs global optimization in the space of structures consisting of atoms,
Historically the first property which USPEX optimized was
enthalpy and search was for stable crystal structures.
Now this target covers atomic and molecular crystals in various dimensions and environments.
Typical target block in this mode looks like
{
type: Atomistic
compositionSpace: {symbols : [Mg Al O] blocks: [[4 8 16]]}
conditions: {externalPressure : 100}
}
Besides obligatory compositionSpace block there are number of
optional parameter blocks and standalone parameters.
compositionSpaceDescribes the identity of each type of atom or molecule and specifies the number of species of each type. For details see Section 5.5.1.
cellUtilityProperties of the unit cell. For details see Section 5.5.2.
environmentUtilitySet up environments (such as substrates) used in the calculation. For details see Section 5.5.3.
radialDistributionUtilityProperties for radial distribution fingerprint calculation. For details see Section 5.5.4.
conditionsProperties of environment in general (currently only external pressure). For details see Section 5.5.5
heredityProperties of heredity variation operator. For details see Section 5.5.6
randSymProperties of symmetrical random structure generator. For details see Section 5.5.7
randSymPyXtalProperties of PyXtal random structure generator. For details see Section 5.5.8
randTopProperties of topological random structure generator. For details see Section 5.5.9
permutationProperties of permutation variation operator. For details see Section 5.5.10
transmutationProperties of transmutation variation operator. For details see Section 5.5.11
softmodemutationProperties of softmutation variation operator. For details see Section 5.5.12
seedsProperties of seeds for the calculation. For details see Section 5.5.13
ionDistances:Sets the minimum interatomic distance matrix between different atom types. Structures with distances lower than
ionDistanceswill be strictly discarded.
- Default:
the
ionDistancesbetween atom A and B are estimated as \(0.22\times(V_{A}^{1/3}+V_{B}^{1/3})\) but not larger than \(1.2 \text{Å}\), and \(0.45\times(V_{A}^{1/3}+V_{B}^{1/3})\) in molecular calculations, where \(V_A\) and \(V_B\) are default volumes of atom A and B estimated in USPEX.- Format:
Note
If the compound in the example above is MgSiO\(_3\), the matrix reads as follows: the minimum Mg—Mg, Mg—Si, Si—Si and O—O distances allowed in a newly generated structure are 1.0 Å while the minimum Mg—O and Si—O distances are 0.7 Å . You can use this keymatrix to incorporate further system-specific information: e.g., if you know that C atoms prefer to be very far apart and are never closer than 3 Å in your system, you can specify this information. Beware, however, that the larger these minimum distances, the more difficult it is to generate structures fulfilling these constraints (especially for large systems), so strive for a compromise and remember that
ionDistancesmust be much smaller than the actual distances in the crystal: realistic distances will be achieved by structure relaxation. WhationDistancestrick does is to avoid structures which cannot be relaxed correctly. Commonly used computational methods (pseudopotentials, PAW, LAPW, and many parametric forcefields) fail when the interatomic distances are too small.
5.5.1. Composition space¶
Examples:
Fixed-compositioin prediction for \(Mg_4 Al_8 O_{16}\).
{ symbols : [Mg Al O] blocks: [[4 8 16]] }
Fixed-compositioin prediction with two molecules of type
mol_1and two of typemol_2in unit cell.{ symbols : [mol_1 mol_2] blocks: [[2 2]] }
Variable-composition prediction for the \(MgO-Al_{2}O_{3}\) system, where the minimum number of atoms is 8 and the maximum 20.
{ symbols : [Mg Al O] blocks: [[1 0 1], [0 2 3]] minAt : 8 maxAt : 20 }
Variable-composition prediction for the \(MgO-Al_2 O_3\) system, where the minimum number of atoms is 8 and the maximum 20. With additional constraint that block \(Al_2 O_3\) should be present in system minimum 1 and maximum 3 times.
{ symbols : [Mg Al O] blocks: [[1 0 1], [0 2 3]] ranges: [(0,10), (1,3)] minAt : 2 maxAt : 20 }
Variable-composition prediction for the \(MgO-Al_2 O_3\) system. Block \(MgO\) should be present in system minimum 2 and maximum 5 times. Block \(Al_2 O_3\) should be present in system minimum 1 and maximum 3 times.
{ symbols : [Mg Al O] blocks: [[1 0 1], [0 2 3]] ranges: [(2,5), (1,3)] }
symbolsDescribes the identity of each type of atom or molecule. The value is a list of chemical elements or molecule names.
Default: no default
- Format:
Molecule names should be defined in molecule definition sections Section 5.9.
blocksSpecify compositional building blocks.
Default: no default
- Format:
rangeSpecify ranges for each block.
Default: If
maxAtis set then each block can be taken from 0 tomaxAtdevided by size of block, otherwise each block is taken one time.
- Format:
maxAtMaximal number of atoms.
Default: If
rangeis set, then maximum of upper bound inrangefor each block times size of block.
- Format:
minAtMinimal number of atoms.
Default: If
rangeis set, then sum of lowest bound inrangefor each block times size of block.
- Format:
5.5.2. Unit cell properties¶
cellVolumeInitial volume of the unit cell.
Default: for cell volumes you don’t have to specify values — USPEX has a powerful algorithm to make reasonable estimates at any pressure.
- Format:
Note
This volume is only used as an initial guess to speed up structure relaxation and does not affect the results, because each structure is fully optimized and adopts the volume corresponding to the (free) energy minimum.
Note
You can also use online program https://uspex-team.org/online_utilities/volume_estimation. Users can also input the volumes manually.
Note
If you study molecular crystals under pressure, you might sometimes need to increase the initial volumes somewhat, in order to be able to generate initial random structures.
cellVectorsUnit cell vectors.
Note
These are used in fixed-cell calculations.
Note
You should provide number of cell vectors equal to dimensionality of the problem.
Default: by default cell vectors are variable.
- Format:
cellParametersUnit cell vectors.
Note
This parameter is used in fixed-cell calculations.
Default: by default cell vectors are variable.
- Format:
dimSpecifies unit cell dimension.
Default: 3
- Format:
thicknessThickness of the surface region or film (for 2D), or maximum linear size of nanoparticle (0D)
Default: 2.0 \(\text{Å}\)
- Format:
supercellDegreeMaximum multiplications of the surface cell, to allow for complex reconstructions.
Default: 1
- Format:
axisVector normal to the plane of the 2D structure.
Default:
- Format:
5.5.3. Environment utility¶
environmentsList of environments. Names used here should be defined in the corresponding definition sections, as described in Section 5.10. Every individual created through random structure generation gets an environment picked up from this list. Individuals created through other variation operators inherit their environments from parents.
- Format:
5.5.4. Radial distribution based fingerprint¶
For details on fingerprint functions we refer reader to Ref 19.
RmaxDistance cuttoff (in Ångstroms).
Default: 10.0
- Format:
deltaDiscretization (in Ångstoms) of the fingerprint.
Default: 0.08
- Format:
sigmaGaussian broadening of interatomic distances (in Ångstoms).
Default: 0.03
- Format:
toleranceSpecifies the minimum cosine distances between structures that qualify them as non-identical — for participating in the production of child structures and for survival of the fittest, respectively. This depends on the precision of structure relaxation and the physics of the system (for instance: for alloy ordering problems, fingerprints belonging to different structures will be very similar, and these tolerance parameters should be made small).
Default: 0.008
- Format:
5.5.5. Conditions¶
externalPressureSpecifies external pressure at which you want to find structures, in GPa.
Default: 0.0
- Format:
Note
Please: do not specify it in relaxation files in the
Specificfolder.
5.5.6. Heredity¶
nslabsHeredity operator might be done traditionally, when both parent structures cut into two pieces. or in ‘zebra’ way. In this case
nslabsdetermines number of slabs into which parents are cut.
- Default:
2 for fixed composition. For variable composition we take length of structure in the direction orthogonal to the cut and divide it by average diameter of atom (double covalent radius).
- Format:
5.5.7. Symmetrical random generator¶
nsymPossible symmetry groups for symmetric random structure generator for crystals (space groups), layer groups for 2D-crystals, wallpaper groups for surfaces, or point groups for clusters. A certain number of structures will be produced using randomly selected groups from this list, using randomly generated lattice parameters and atomic coordinates. During this process special Wyckoff sites can be produced from general positions (Fig. 5.5.1, see Ref. 14 for details)
- Default:
For 3D crystals: 2-230
- Format:
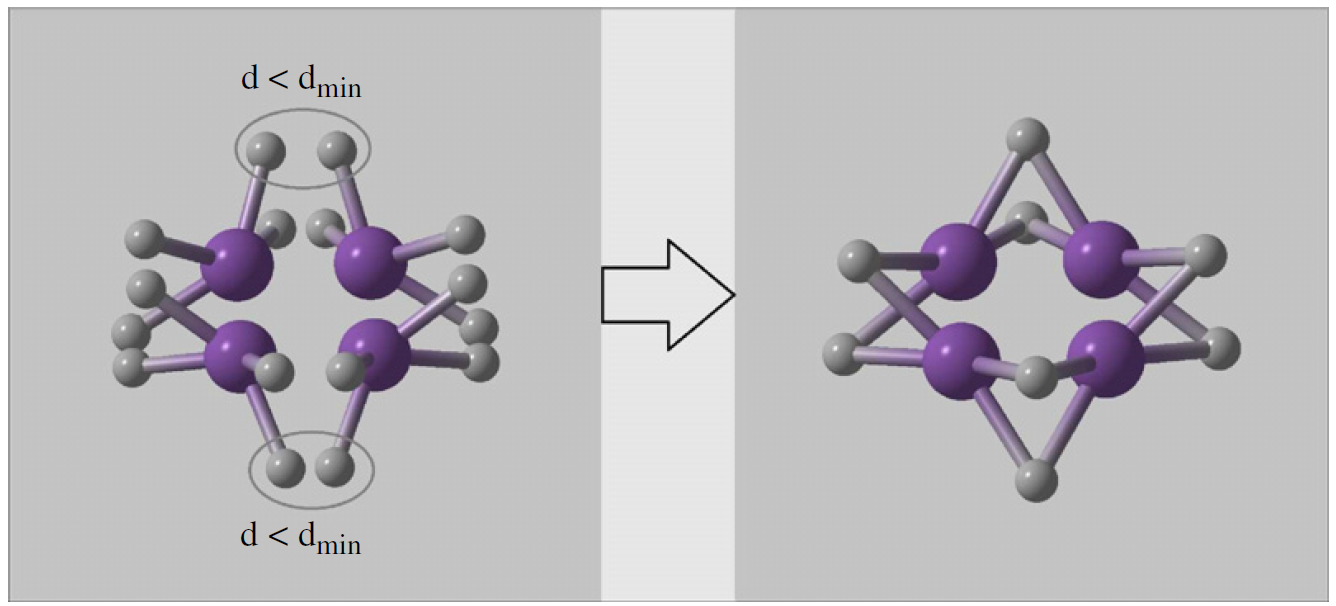
Fig. 5.5.1 Example of random symmetric structure generation and merging atoms onto special Wyckoff positions.¶
splitIntoDefines the number of identical subcells or pseudosubcells in the unit cell. If you do not want to use splitting, just use the value 1, or delete the block. Use splitting only for systems with >25-30 atoms/cell.
Default: 1
- Format:
Subcells introduce extra translational (pseudo)symmetry. In addition to this, each subcell can be built using symmetric random structure generator developed by A.R. Oganov and H.T. Stokes and implemented by H.T. Stokes (see Reference 14).
5.5.8. RandSymPyXtal¶
nsymPossible symmetry groups for symmetric random structure generator for crystals (space groups), layer groups for 2D-crystals, wallpaper groups for surfaces, or point groups for clusters. A certain number of structures will be produced using randomly selected groups from this list, using randomly generated lattice parameters and atomic coordinates. During this process special Wyckoff sites can be produced from general positions (Fig. 5.5.1)
- Default:
For 3D crystals: 2-230
- Format:
5.5.9. Topological random generator¶
maxSupersizeWhen trying to generate structure with certain number of atoms topological random generator takes from database of topologies all entries number of nodes in which fits the required one up to the factor of
maxSupersize.
- Default:
4
- Format:
supercellsList of allowed supercells. When trying to generate structure with certain number of atoms topological random generator takes from database of topologies all entries which when replicated according
supercellsgive required number of atoms.
- Default:
all supercells are allowed, up to
maxSupersize.- Format:
5.5.10. Permutation¶
howManySwapsFor permutation, the number of pairwise swaps will be randomly drawn from a uniform distribution between 1 and
howManySwaps.
- Default:
0.5\(\times\)(maximum number of possible swaps). If atoms \(N_a\) and \(N_b\), and atoms \(N_c\) and \(N_d\) are swappable, then the total number of possible swaps is \(\min(N_a,N_b)+\min(N_c,N_d)\), and the default for is \(0.5\times[\min(N_a,N_b)+\min(N_c,N_d)]\). In most cases, it is a good idea to rely on this default.
- Format:
specificSwapsSpecifies which atom types you allow to swap in permutation.
Default: No specific swaps and all atoms are permutable
- Format:
Note
In this case, atoms of type 1 can be swapped with atoms of type 2. If you want to try all possible swaps, just leave a blank line inside this keyblock, or delete the block.
5.5.11. Transmutation¶
In this operator, a randomly selected atom is transmuted into another chemical species present in the system - the new chemical identity is chosen randomly.
howManyTransMaximum percentage of atoms in the structure that are being transmuted (0.1 = 10%). The fraction of atoms that will be transmuted is drawn randomly from a homogeneous distribution bounded from 1 to the fractional parameter
howManyTrans.Default: 0.2
- Format:
5.5.12. Softmutation¶
degreeThe maximum displacement in softmutation in angstoms. The displacement vectors for softmutation are scaled so that the largest displacement magnitude equals
degree.Default: 3\(\times\)(average atomic radius)degree: 1.0
- Format:
5.5.13. Seeds¶
This feature requires aditional input files to be provided.
generationsList of generations into which seeds will be injected during the search.
Default: No default
- Format:
seedsFoldersList of paths to folders which contain seeds files to be injected during the search. In the same order as generations above.
Default:
- Format:
5.6. Evolutionary algorithm USPEX¶
When doing global optimization search (see Section 5.3) one can choose between several algorithms. In current release only evolutionary algorithm is implemented – which is the most efficient and reliable one. Particle swarm optimization and evolutionary metadynamics will be made available soon.
popSizeThe number of structures in each generation; size of initial generation can be set separately, if needed.
Default: \(2 \times N\) rounded to the closest 10, where \(N\) is the number of atoms/cell (or for variable composition). The upper limit is 60. Usually, you can trust these default settings. popSize: 20
- Format:
initialPopSizeThe number of structures in the initial generation.
Default: equal to
populationSize
- Format:
Note
In most situations, we suggest that these two parameters be equal.
Sometimes (especially in variable-composition calculations)
it may be useful to specify initialPopSize to be larger
than populationSize. It is also possible to have
a smaller initialPopSize, if one wants to
produce the first generation from seed structures.
bestFracFraction of the current generation that shall be used as potential parents to produce the next generation.
Default: 0.7
- Format:
Note
This is an important parameter, values between 0.5–0.8 are reasonable.
howManyDiverseDefines how many good and diverse structures will survive into the next generation.
Default: 0.15\(\times\)
popSize
- Format:
We perform clustering of the population into specified number of groups here. And take the fittest representative from each group
optTypeThis keyblock specifies the property that you wish to use for selecting potential parents for new generation. See Section 5.4.
Default: same as in global optimizer section.
- Format:
Fittness can differ from the property to be optimized, if one uses aging procedure.
fractionsThis parameter defines allowed percentages of each variation operator. The first value is minimum percentage of structures in each generation produced by this operator. The second — maximum percentage. The third value is weight (not the percentage) of the operator in the 1st generation.
- Format:
The fractions of operators evolve during the calculation, so that the more successful operators gain weight at the expense of the less successful operators, but within the limits specified here.
5.7. Details of ab initio calculations sections¶
Typical ab initio definition section looks like
#define vasp1
{
type: vasp
commandExecutable: 'vasp'
kresol : 0.12
vacuumSize: 10 #only for 0- 1- or 2- dimensional calculations
}
It contains type of interface for external program package, common
parameters (like commandExecutable,
see Section 5.7.1), package specific
parameters (like kresol) and vacuumSize which should be specified only
for low-dimensional systems and should be not specified for 3D-crystals.
The definiton name (vasp1 in the example above) should be used
in \(stages\) parameter of general parameters
Section 5.2.
Available types of interfaces are:
vasp— VASP interface, see Section 5.7.2.gulp— GULP interface, see Section 5.7.3.lammps— LAMMPS interface, see Section 5.7.4.qe— Quantum Espresso interface, see Section 5.7.5.abinit— Abinit interface, see Section 5.7.6.aims— FHIaims interface, see Section 5.7.7.mopac— mopac interface, see Section 5.7.8.
5.7.1. Common interface parameters.¶
commandExecutableSpecifies executable for a given code
Default: no default, has to be specified by the user.
- Format:
taskManagerSpecifies name of task manager to be used for submission.
Default: By default no task manager is used.
- Format:
Note
Usually you define one task manager and use it in several stages, but it can be useful to use different task managers for different stages. For details see Section 5.8
tagOptional identifier for stage.
Default: equal to index number of stage.
- Format:
Note
This is useful if you want to highlight or distinguish a stage. This tag will appear
as suffix for a job in job submission system and in calculation folder created for this job. If
this parameter is set then files in Specific/ folder should use it in their suffixes instead of index
number of stage. For eaxample INCAR_final istead of just INCAR_4.
5.7.2. VASP interface parameters.¶
sleepTimeSpecifies sleep delay between checks of completion for this stage in seconds.
Default: 30
- Format:
kresolSpecifies the reciprocal-space resolution for k-points generation.
- Format:
Note
Using different values for each step of structure relaxation, starting with cruder (i.e., larger) values and ending with high resolution dramatically speeds up calculations, especially for metals, where very many k-points are needed. This keyblock is important for ab-initio calculations (through some codes, e.g. VASP and SIESTA, now have similar tricks)).
vacuumSizeSpecify vacuum region size for calculations of nanoparticles, films or surfaces.
Default: 10.0
- Format:
5.7.3. GULP interface parameters.¶
sleepTimeSpecifies sleep delay between checks of completion for this stage in seconds.
Default: 10
- Format:
vacuumSizeSpecify vacuum region size for calculations of nanoparticles, films or surfaces.
Default: 10.0
- Format:
5.7.4. LAMMPS interface parameters.¶
sleepTimeSpecifies sleep delay between checks of completion for this stage in seconds.
Default: 10
- Format:
vacuumSizeSpecify vacuum region size for calculations of nanoparticles, films or surfaces.
Default: 10.0
- Format:
5.7.5. Quantum Espresso interface parameters.¶
sleepTimeSpecifies sleep delay between checks of completion for this stage in seconds.
Default: 30
- Format:
kresolSpecifies the reciprocal-space resolution for k-points generation.
- Format:
Note
Using different values for each step of structure relaxation, starting with cruder (i.e., larger) values and ending with high resolution dramatically speeds up calculations, especially for metals, where very many k-points are needed. This keyblock is important for ab-initio calculations (through some codes, e.g. VASP and SIESTA, now have similar tricks)).
vacuumSizeSpecify vacuum region size for calculations of nanoparticles, films or surfaces.
Default: 10.0
- Format:
5.7.6. Abinit interface parameters.¶
sleepTimeSpecifies sleep delay between checks of completion for this stage in seconds.
Default: 30
- Format:
kresolSpecifies the reciprocal-space resolution for k-points generation.
Default:
- Format:
Note
Using different values for each step of structure relaxation, starting with cruder (i.e., larger) values and ending with high resolution dramatically speeds up calculations, especially for metals, where very many k-points are needed. This keyblock is important for ab-initio calculations (through some codes, e.g. VASP and SIESTA, now have similar tricks)).
vacuumSizeSpecify vacuum region size for calculations of nanoparticles, films or surfaces.
Default: 10.0
- Format:
5.7.7. FHIaims interface parameters.¶
sleepTimeSpecifies sleep delay between checks of completion for this stage in seconds.
Default: 30
- Format:
kresolSpecifies the reciprocal-space resolution for k-points generation.
Default:
- Format:
5.7.8. MOPAC interface parameters.¶
sleepTimeSpecifies sleep delay between checks of completion for this stage in seconds.
Default: 1
- Format:
5.8. Task manager definition¶
Task manager definition looks like
#define TM
{
type: SBATCH
header: "#!/bin/sh
#SBATCH -p debug
#SBATCH -N 1
#SBATCH -n 1
"
}
Type could be:
SBATCH— for slurm submission system,QSUB— for torque submission system,BSUB— for lsf submission system.
Header should contain fraction of submission script for supercomputer which you are going to use. Usually such header should contain information on submission queue, number of nodes and cores which the job is going to consume. To determine exact content of such header consult with supercomputer usage guide or administrator.
5.9. Molecules definitions¶
Each molecule definition looks like
#define mol_name
{filename : 'MOL_FILE'}
The defined molecule name (like \(mol\_name\)) should then be used
in compositionSpace block (see Section 5.5.1).
For a molecular crystal, the MOL_FILE file describes the internal geometry of the
molecule from which the structure is built. The Z Matrix file is created using
the information given in the MOL_FILE file, i.e., bond lengths and all
necessary angles are calculated from the Cartesian coordinates. The
lengths and angles that are important should be used for the creation of Z Matrix
— this is exactly what columns 5–7 specify. Let’s look at the file for
benzene \(C_6 H_6\):
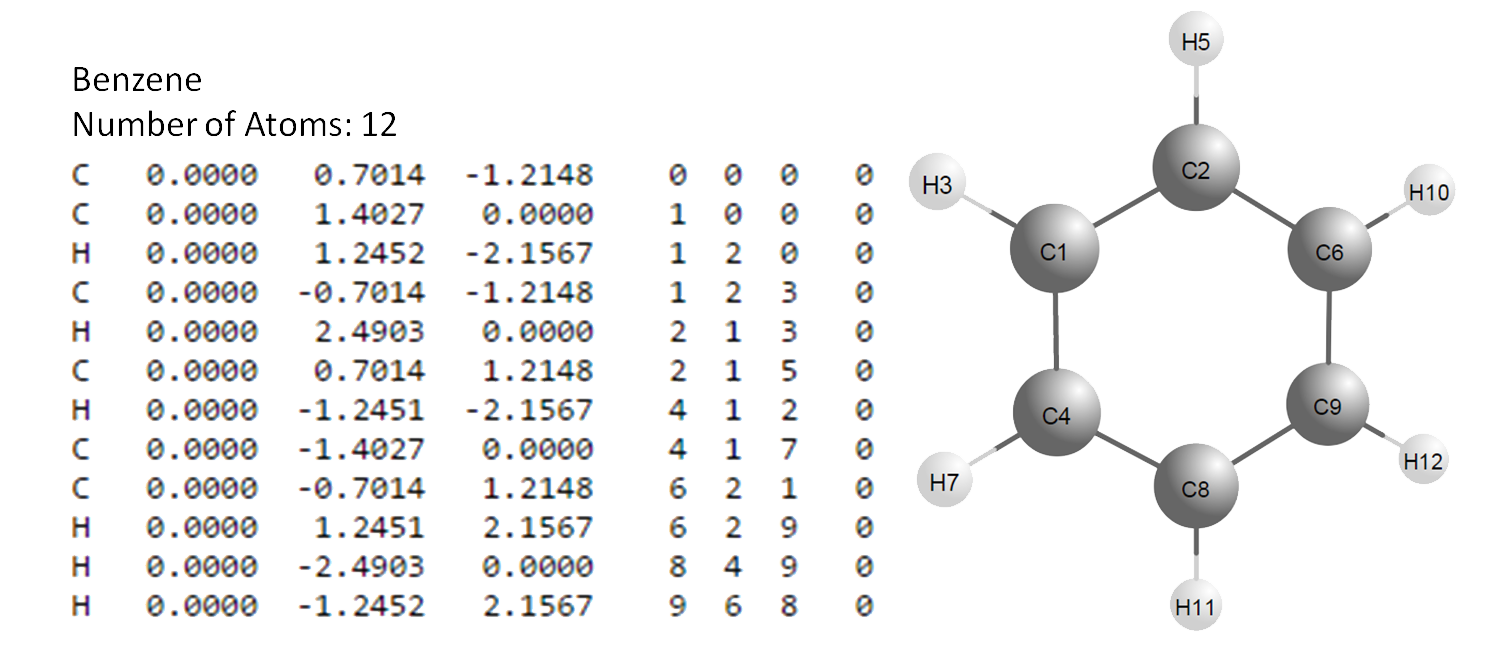
Fig. 5.9.1 Sample of MOL_FILE file and illustration of the corresponding molecular
structure.¶
The 1\(^{st}\) atom is C, its coordinates are defined without reference to other atoms (“0 0 0”).
The 2\(^{nd}\) atom is C, its coordinates (in molecular coordinate
frame) in Z Matrix will be set only by its distance from the 1\(^{st}\)
atom (i.e. C described above), but no angles — (“1 0 0”).
The 3\(^{rd}\) atom is H, its coordinates will be set by its distance from the 1\(^{st}\) atom, and the bond angle 3-1-2, but not by torsion angle — hence we use “1 2 0”.
The 4\(^{th}\) atom is C, its coordinates will be set by its distance from the 1\(^{st}\) atom, bond angle 4-1-2, and torsion angle 4-1-2-3 — hence, we use “1 2 3” and so forth…until we reach the final, 12\(^{th}\) atom, which is H, defined by its distance from the 9\(^{th}\) atom (C), bond angle 12-9-6 and torsion angle 12-9-6-8 — hence “9-6-8”.
The final column is the flexibility flag for the torsion angle. For example, in C4, the tosion angle is defined by 4-1-2-3. This flag should be either 1 or 0 for the first three atoms, and 0 — for the others, if the molecule is rigid. If any other flexible torsion angle exists, specify 1 for this column.
5.9.1. How to prepare the MOL files¶
There are plenty of programs which can generate Zmatrix style files, such as Molden, Avogadro, and so on. Experienced users might have their own way to prepare these files. For the users’ convenience, we have created an online utility to allow one to generate the USPEX-style MOL file just from a file in XYZ format. Please try this utility at https://uspex-team.org/online_utilities/zmatrix/
5.10. Environment definitions¶
Environment definition looks like.
#define substrate
{
type: substrate
file: './POSCAR_SUBSTRATE'
pbc: (1 1 0)
bufferThickness: 3.0
}
The only supported type now is substrate.
Here file should contain substrate region in VASP5 POSCAR format, see Fig. 5.10.1.
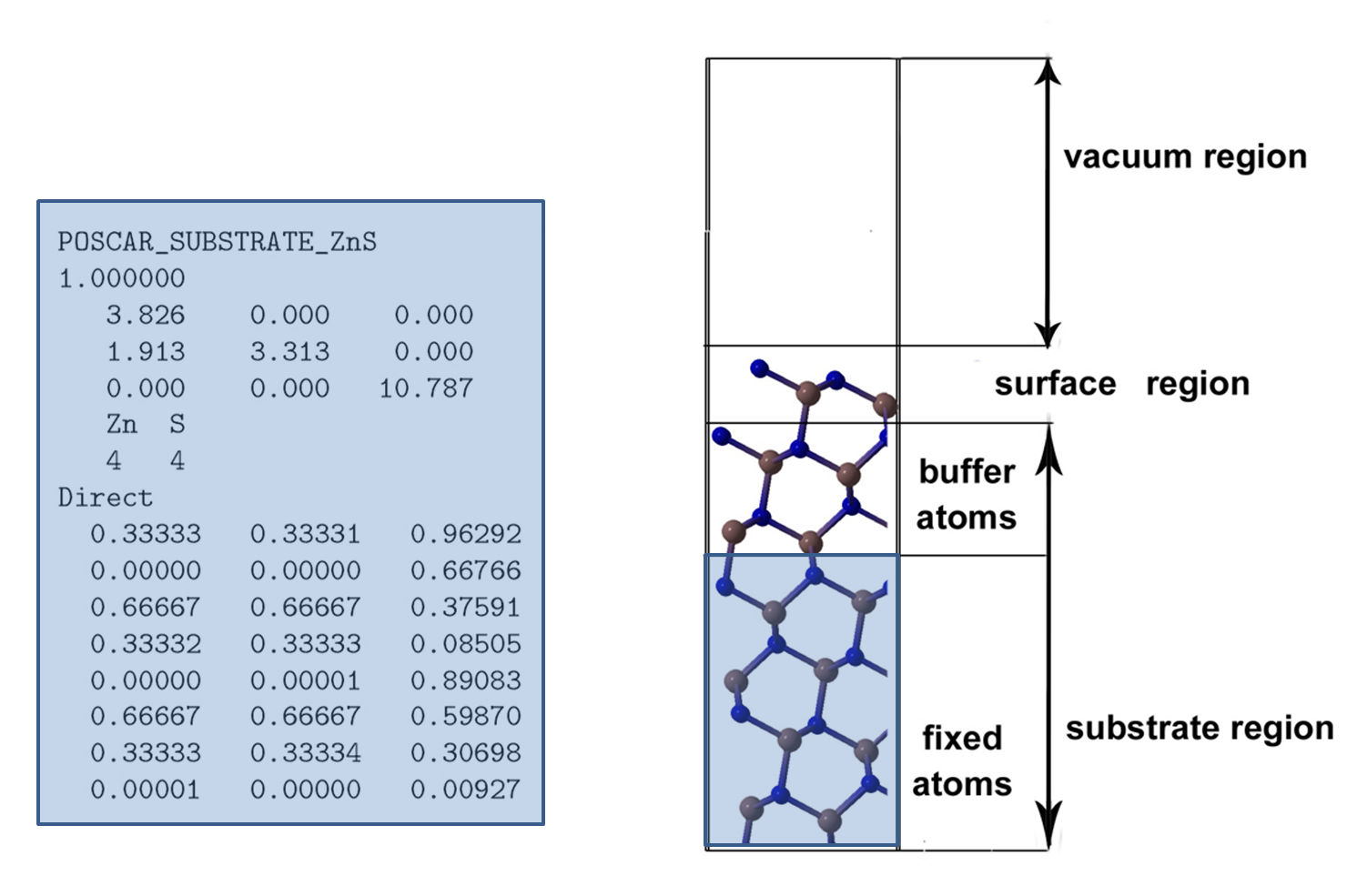
Fig. 5.10.1 Surface model used in USPEX.¶
Besides file you need to specify following parameters.
pbc: (1 1 0)Periodic boundary conditions. 1 indicates the persistence of periodic boundary conditions and 0 indicates vacuum direction
- Format:
bufferThicknessThickness of the buffer region in substrate. This region is part of
POSCAR_SUBSTRATE, and is allowed to relax. See Fig. 5.10.1.
- Format: