为了完成表面重构的预测,必须:
指定 200 或 201 : 计算类型。
准备包含基底的VASP5 POSCAR格式的文件,如图11 10所示。
列举以下参数:
![\includegraphics[scale=0.5]{pic/Surface.png}](images/img-0103.png)
% symmetries 2-17 % endSymmetries
注意: 如果对称标签symmetries出现,USPEX将尝试使用平面组生成结构。
 variable thicknessS
variable thicknessS
Meaning: 表面区域的厚度。只有这个区域允许吸附原子。
Default: 2.0 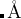
Format:
3.5 : thicknessS
variable thicknessB
Meaning: 基底的缓冲区域的厚度。这个区域是POSCAR_SUBSTRATE的一部分, 可以松弛。
Default: 3.0
Format:
3.0 : thicknessB
variable reconstruct
Meaning: 表面单元的最大增值,允许复杂重建。
Default: 1
Format:
1 : reconstruct
USPEX也提供研究表面原子变成分搜索的可能(在USPEX 10.1发布)。在这种情况下, 稳定的表面重建是由化学势控制的。典型的输入设置如下:
****************************************** * TYPE OF RUN AND SYSTEM * ****************************************** USPEX : calculationMethod (USPEX, VCNEB, META) 201 : calculationType (dimension: 0-3; molecule: 0/1; varcomp: 0/1) % atomType Si O % EndAtomType % numSpecies 2 4 % EndNumSpecies
这里我们列出 晶胞表面原子的最大数。
晶胞表面原子的最大数。
****************************************** * SURFACES * ****************************************** % symmetries 2-17 % endSymmetries % StoichiometryStart 1 2 % StoichiometryEnd
这定义晶体的化学计量数。
-23.7563 : E_AB (AmBn化合物的DFT能量,eV/化学式)
-5.4254 : Mu_A (元素A的DFT能量, eV/原子)
-4.9300 : Mu_B (元素B的DFT能量, in eV/atom)
3.5 : thicknessS (表面区域的厚度, 默认2 $\text{\r{A}}$ )
3.0 : thicknessB (基底缓冲区域的厚度, 默认3 $\text{\r{A}}$)
4 : reconstruct (晶胞的最大倍数)
现在,USPEX支持如下的表面变成分计算:
基本的表面重建(如C在diamond(100) 表面)。
二元化合物的表面重建(如GaN(0001) 表面)。
包含不同物种的基本表面的重建(如PdO在Pd(100)表面)。