| USPEX 9.4.4 manual |
| USPEX 9.4.4 manual |
This technique is useful, if instead of starting with random structures, you would like to input some structures that you already know for the compound or related materials. Just create a file Seeds/POSCARS for the next generation, or Seeds/POSCARS_gen (gen is the generation number) for the specific generation of an USPEX calculation, in the format of concatenated POSCAR files in VASP5 format. Don’t miss letter “S” in the file name.
Example:
EA33 2.69006 5.50602 4.82874 55.2408 73.8275 60.7535 no SG
1.0
2.690100 0.000000 0.000000
2.690100 4.804100 0.000000
1.344900 2.402100 3.967100
Mg Al O
1 2 4
Direct
0.799190 0.567840 0.859590
0.793520 0.230950 0.544750
0.793540 0.916090 0.174450
0.050972 0.816060 0.859610
0.172230 0.194810 0.859600
0.438250 0.655170 0.406880
0.438230 0.202440 0.312330
EA34 7.61073 2.85726 2.85725 60.0001 79.1809 79.1805 no SG
1.0
7.610700 0.000000 0.000000
0.536350 2.806500 0.000000
0.536330 1.352000 2.459300
Mg Al O
1 2 4
Direct
0.708910 0.507440 0.068339
0.374050 0.285730 0.846630
0.023663 0.069185 0.630090
0.889560 0.780560 0.341460
0.350470 0.626920 0.187820
0.597290 0.211310 0.772210
0.116440 0.371590 0.932500
One can add seeds at any time during the calculation. USPEX will look for new seeds at the beginning of each generation. The corresponding information will be recorded to results1/Seeds_history and the seeds files (POSCARS or POSCARS_gen) will be kept as POSCARS_gen in Seeds/ folder.
Whenever seeds are added, we suggest users to check the results1/Seeds_history and Warnings files. There will be a warning message “Meet a problem when reading Seeds - ...” if your seeds are problematic. When an error appears in the seeds file, such as missing lines, the structures after the error point will not be added.
Note: Make sure you specified all atomic symbols at the 6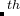 line of each structure. For example, to add the
line of each structure. For example, to add the 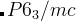 H
H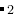 structure to a H-O variable-composition calculation, you should edit the file as:
structure to a H-O variable-composition calculation, you should edit the file as:
H_I-P63/mc
1
4.754726 -2.74514 0.000000
-0.00000 5.490285 0.000000
0.000000 0.000000 4.508715
H
16
Direct
...
(the atomic positions information is omitted here)
| USPEX 9.4.4 manual |